Chapter Headings
Key Messages
- Diabetic ketoacidosis and hyperosmolar hyperglycemic state should be suspected in people who have diabetes and are ill. If either diabetic ketoacidosis or hyperosmolar hyperglycemic state is diagnosed, precipitating factors must be sought and treated.
- Diabetic ketoacidosis and hyperosmolar hyperglycemic state are medical emergencies that require treatment and monitoring for multiple metabolic abnormalities and vigilance for complications.
- A normal or mildly elevated blood glucose level does not rule out diabetic ketoacidosis in certain conditions, such as pregnancy or with SGLT2 inhibitor use.
- Diabetic ketoacidosis requires intravenous insulin administration (0.1 units/kg/h) for resolution. Bicarbonate therapy may be considered only for extreme acidosis (pH ≤7.0).
Key Messages for People with Diabetes
When you are sick, your blood glucose levels may fluctuate and be unpredictable:
- During these times, it is a good idea to check your blood glucose levels more often than usual (for example, every 2 to 4 hours).
- Drink plenty of sugar-free fluids or water.
- If you have type 1 diabetes with blood glucose levels remaining over 14 mmol/L before meals, or if you have symptoms of diabetic ketoacidosis (see Table 1
- Develop a sick-day plan with your diabetes health-care team. This should include information on:
-
Which diabetes medications you should continue and which ones you should temporarily stop
-
Guidelines for insulin adjustment if you are on insulin
-
Advice on when to contact your health-care provider or go to the emergency room.
-
Note: Although the diagnosis and treatment of diabetic ketoacidosis (DKA) in adults and in children share general principles, there are significant differences in their application, largely related to the increased risk of life-threatening cerebral edema with DKA in children and adolescents. The specific issues related to treatment of DKA in children and adolescents are addressed in the Type 1 Diabetes in Children and Adolescents chapter, p. S234.
Introduction
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) are diabetes emergencies with overlapping features. With insulin deficiency, hyperglycemia causes urinary losses of water and electrolytes (sodium, potassium, chloride) and the resultant extracellular fluid volume (ECFV) depletion. Potassium is shifted out of cells, and ketoacidosis occurs as a result of elevated glucagon levels and insulin deficiency (in the case of type 1 diabetes). There may also be high catecholamine levels suppressing insulin release (in the case of type 2 diabetes). In DKA, ketoacidosis is prominent while, in HHS, the main features are ECFV depletion and hyperosmolarity. HHS is the preferred term to describe this condition as opposed to hyperosmolar nonketotic coma (HONKC) since less than one-third of people with HHS actually present with a coma (1).
Risk factors for DKA include new diagnosis of diabetes mellitus, insulin omission, infection, myocardial infarction (MI), abdominal crisis, trauma and, possibly, continuous subcutaneous insulin infusion (CSII) therapy, thyrotoxicosis, cocaine, atypical antipsychotics and, possibly, interferon. HHS is much less common than DKA (2,3). In addition to the precipitating factors noted above for DKA, HHS also has been reported following cardiac surgery and with the use of certain drugs, including diuretics, glucocorticoids, lithium and atypical antipsychotics. Infections are present in 40% to 60% of people with HHS (4). In up to 20% of cases of HHS, individuals had no prior history of diabetes (4).
The clinical presentation of DKA includes symptoms and signs of hyperglycemia, acidosis and the precipitating illness (Table 1). In HHS, there is often more profound ECFV contraction and decreased level of consciousness (proportional to the elevation in plasma osmolality). In addition, in HHS, there can be a variety of neurological presentations, including seizures and a stroke-like state that can resolve once osmolality returns to normal (3,5,6). In HHS, there also may be evidence of a precipitating condition similar to DKA.
In individuals with type 2 diabetes, the incidence of DKA is estimated to be in the range of 0.32 to 2.0 per 1,000 patient-years (7) while, in people with type 1 diabetes, the incidence is higher at 4.6 to 8.0 per 1000 patient-years (8). There is a group of individuals with diabetes that present with DKA but do not have the typical features of type 1 diabetes. There are various terms given to characterize this condition, such as flatbush diabetes, type 1.5 diabetes, atypical diabetes or type 1B diabetes, but it may be most useful to label this state as ketosis-prone diabetes (KPD). There are several classification systems used to describe KPD that take into account pathophysiology and prognosis. Individuals with KPD have very little beta cell function, may or may not have beta cell antibodies, and some may require temporary or lifelong insulin therapy (9).
| Table 1 Clinical presentation of DKA |
||
|---|---|---|
| Symptoms | Signs | |
| Hyperglycemia | Polyuria, polydipsia, weakness | ECFV contraction |
| Acidosis | Air hunger, nausea, vomiting and abdominal pain Altered sensorium |
Precipitating condition |
| Precipitating condition | See list of conditions in Table 2 | |
Prevention
Sick-day management that includes capillary beta-hydroxybutyrate monitoring reduces emergency room visits and hospitalizations in young people (10).
SGLT2 Inhibitors and DKA
SGLT2 inhibitors may lower the threshold for developing DKA through a variety of different mechanisms (11–13). The presentation of the DKA is similar to those who develop DKA without SGLT2 inhibitor exposure, except that the blood glucose (BG) levels on presentation may not be as elevated as expected. In randomized controlled trials, the incidence of DKA associated with SGLT2 inhibitors is low (≤0.1% of treated people) (14,15). In most cases, there is usually a known precipitant as a contributing factor, such as insulin dose reduction or omission, bariatric surgery or other surgery, alcohol, exercise, or low carbohydrate or reduced food intake (16–20).
Diagnosis
DKA or HHS should be suspected whenever people have significant hyperglycemia, especially if they are ill or highly symptomatic (see above). As outlined in Figure 1
There are no definitive criteria for the diagnosis of DKA. Typically, the arterial pH is ≤7.3, serum bicarbonate is ≤15 mmol/L and the anion gap is >12 mmol/L with positive serum and/or urine ketones (1,31–33). PG is usually ≥14.0 mmol/L but can be lower, especially with the use of SGLT2 inhibitors (34). DKA is more challenging to diagnose in the presence of the following conditions: 1) mixed acid-base disorders (e.g. associated vomiting, which will raise the bicarbonate level); 2) if there has been a shift in the redox potential, favouring the presence of beta-OHB (rendering serum ketone testing negative); or 3) if the loss of keto anions with sodium or potassium in osmotic diuresis has occurred, leading to a return of the plasma anion gap toward normal. It is, therefore, important to measure ketones in both the serum and urine. If there is an elevated anion gap and serum ketones are negative, beta-OHB levels should be measured. Negative urine ketones should not be used to rule out DKA (35).
Measurement of serum lactate should be considered in hypoxic states. In HHS, a more prolonged duration of relative insulin insufficiency and inadequate fluid intake (or high glucose intake) results in higher PG levels (typically ≥34.0 mmol/L), plasma osmolality >320 mOsm/kg and greater ECFV contraction, but minimal acid-base disturbance (1,31).
Pregnant women in DKA typically present with lower PG levels than nonpregnant women (36), and there are case reports of euglycemic DKA in pregnancy (37,38).
| Table 2 Priorities* to be addressed in the management of adults presenting with hyperglycemic emergencies |
||
|---|---|---|
| DKA, diabetic ketoacidosis; ECFV, extracellular fluid volume; HHS, hyperosmolar hyperglycemic state. Severity of issue will dictate priority of action. |
||
| Metabolic | Precipitating cause of DKA/HHS | Other complications of DKA/HHS |
|
|
|
Management
Objectives of management include restoration of normal ECFV and tissue perfusion; resolution of ketoacidosis; correction of electrolyte imbalances and hyperglycemia; and the diagnosis and treatment of coexistent illness. The issues that must be addressed in the individual presenting with DKA or HHS are outlined in Table 2
People with DKA and HHS are best managed in an intensive care unit or step-down setting (1,31,32) with specialist care (39,40). Protocols and insulin management software systems (41) may be beneficial (42,43), but there can be challenges with achieving adherence (44,45). Volume status (including fluid intake and output), vital signs, neurological status, plasma concentrations of electrolytes, anion gap, osmolality and glucose need to be monitored closely, initially as often as every 2 hours (1,31,32). Capillary blood glucose (CBG) measurements are unreliable in the setting of severe acidosis (46). Precipitating factors must be diagnosed and treated (1,31,32).
Extracellular fluid volume contraction
The sodium deficit is typically 7 to 10 mmol/kg in DKA (47) and 5 to 13 mmol/kg in HHS, which, along with water losses (100 mL/kg and 100 to 200 mL/kg, respectively), results in decreased ECFV, usually with decreased intracellular fluid volume (47). Restoring ECFV improves tissue perfusion and reduces plasma glucose levels both by dilution and by increasing urinary glucose losses. ECFV re-expansion, using a rapid rate of initial fluid administration, was associated with an increased risk of cerebral edema in 1 study (48) but not in another (49). In adults, one should initially administer intravenous normal saline 1 to 2 L/h to correct shock, otherwise 500 mL/h for 4 hours, then 250 mL/h of intravenous fluids (50,51).
Figure 1
Management of diabetic ketoacidosis in adults.
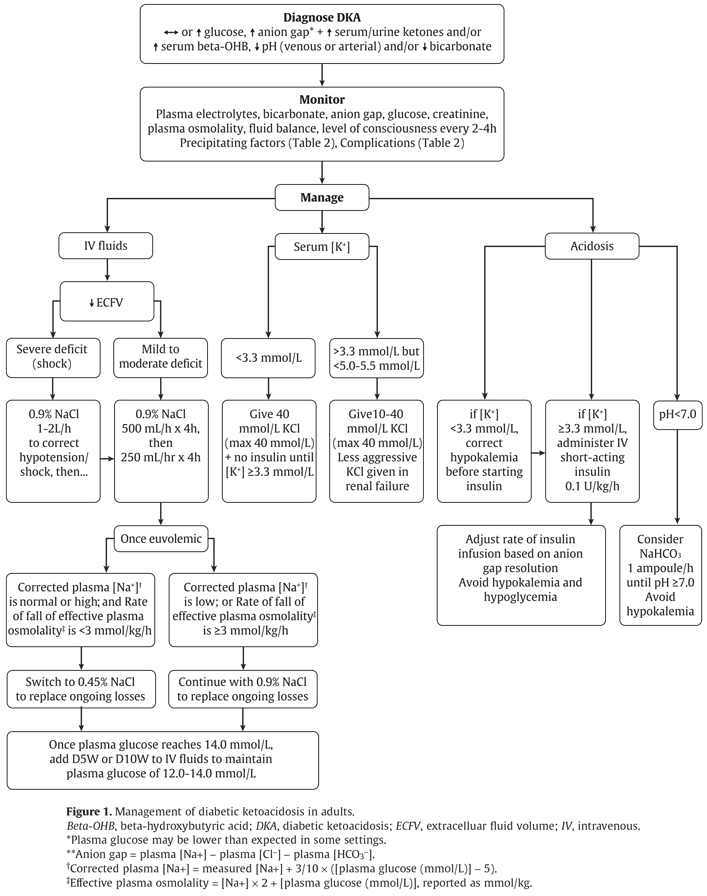
Beta-OHB, beta-hydroxybutyric acid; DKA, diabetic ketoacidosis; ECFV, extracelluar fluid volume; IV, intravenous.
*Plasma glucose may be lower than expected in some settings.
**Anion gap = plasma [Na+] − plasma [Cl
†Corrected plasma [Na+] = measured [Na+] + 3/10 × ([plasma glucose (mmol/L)] − 5).
‡Effective plasma osmolality = [Na+] × 2 + [plasma glucose (mmol/L)], reported as mmol/kg.
Potassium deficit
The typical potassium deficit range is 2 to 5 mmol/kg in DKA and 4 to 6 mmol/kg in HHS (48). There have been no randomized trials that have studied strategies for potassium replacement. Typical recommendations suggest that potassium supplementation should be started for plasma potassium <5.0 to 5.5 mmol/L once diuresis has been established, usually with the second litre of saline. If the individual at presentation is normo- or hypokalemic, potassium should be given immediately, at concentrations in the intravenous fluid between 10 to 40 mmol/L, at a maximum rate of 40 mmol/h.
In the case of frank hypokalemia (serum potassium <3.3 mmol/L), insulin should be withheld until potassium replacement at 40 mmol/h has restored plasma potassium to ≥3.3 mmol/L (1,31). It is reasonable to treat the potassium deficit of HHS in the same way.
| Table 3 Summary of fluid therapy for DKA and HHS in adults |
|---|
| DKA, diabetic ketoacidosis; HHS, hyperosmolar hyperglycemic state; IV, intravenous. |
|
Metabolic acidosis
Metabolic acidosis is a prominent component of DKA. People with HHS have minimal or no acidosis. Insulin is used to stop ketoacid production; intravenous fluid alone has no impact on parameters of ketoacidosis (52). Short-acting insulin (0.1 units/kg/h) is recommended (53–55). There is no conclusive evidence supporting the use of an initial insulin bolus in adults and it is not recommended in children. Although the use of an initial bolus of intravenous insulin is recommended in some reviews (1), there has been only 1 randomized controlled trial in adults examining the effectiveness of this step (56). In this study, there were 3 arms: a bolus arm (0.07 units/kg, then 0.07 units/kg/h), a low-dose infusion group (no bolus, 0.07 units/kg/h) and a double-dose infusion group (no bolus, 0.14 units/kg/h). Outcomes were identical in the 3 groups, except 5 of 12 participants needed extra insulin in the no-bolus/low-dose infusion group, and the double-dose group had the lowest potassium (nadir of 3.7 mmol/L on average). Unfortunately, this study did not examine the standard dose of insulin in DKA (0.1 units/kg/h). In children, using an initial bolus of intravenous insulin does not result in faster resolution of ketoacidosis (57,58) and increases the risk of cerebral edema (see Type 1 Diabetes in Children and Adolescents chapter, p. S234).
A systematic review based on low- to very-low-quality evidence, showed that subcutaneous hourly analogues provide neither advantages nor disadvantages compared to intravenous regular insulin when treating mild to moderate DKA (59). The dose of insulin should subsequently be adjusted based on ongoing acidosis (60), using the plasma anion gap or beta-OHB measurements.
Use of intravenous sodium bicarbonate to treat acidosis did not affect outcome in randomized controlled trials (61–63). Sodium bicarbonate therapy may be considered in adult individuals in shock or with arterial pH ≤7.0. For example, one can administer 1 ampoule (50 mmol) sodium bicarbonate added to 200 mL D5W (or sterile water, if available) over 1 hour, repeated every 1 to 2 hours, until pH is ≥7.0 (1,31). Potential risks associated with the use of sodium bicarbonate include hypokalemia (64) and delayed occurrence of metabolic alkalosis.
Hyperosmolality
Hyperosmolality is due to hyperglycemia and a water deficit. However, serum sodium concentration may be reduced due to shift of water out of cells. The concentration of sodium needs to be corrected for the level of glycemia to determine if there is also a water deficit (Figure 1). In people with DKA, plasma osmolality is usually ≤320 mmol/kg. In HHS, plasma osmolality is typically >320 mmol/kg. Because of the risk of cerebral edema with rapid reductions in osmolality (65), it has been recommended that the plasma osmolality be lowered no faster than 3 mmol/kg/h (1,31). This can be achieved by monitoring plasma osmolality, by adding glucose to the infusions when PG reaches 14.0 mmol/L to maintain it at that level and by selecting the correct concentration of intravenous saline. Typically, after volume re-expansion, intravenous fluid may be switched to half-normal saline because urinary losses of electrolytes in the setting of osmotic diuresis are usually hypotonic. The potassium in the infusion will also add to the osmolality. If osmolality falls too rapidly despite the administration of glucose, consideration should be given to increasing the sodium concentration of the infusing solution (1,31). Water imbalances can also be monitored using the corrected plasma sodium. Central pontine myelinolysis has been reported in association with overly rapid correction of hyponatremia in HHS (66).
PG levels will fall due to multiple mechanisms, including ECFV re-expansion (67), glucose losses via osmotic diuresis (52), insulin-mediated reduced glucose production and increased cellular uptake of glucose. Once PG reaches 14.0 mmol/L, intravenous glucose should be started to prevent hypoglycemia, targeting a plasma glucose of 12.0 to 14.0 mmol/L. Similar doses of intravenous insulin can be used to treat HHS, although these individuals are not acidemic, and the fall in PG concentration is predominantly due to re-expansion of ECFV and osmotic diuresis (67). Insulin has been withheld successfully in HHS (68), but generally its use is recommended to reduce PG levels (1,31).
Phosphate deficiency
There is currently no evidence to support the use of phosphate therapy for DKA (69–71), and there is no evidence that hypophosphatemia causes rhabdomyolysis in DKA (72). However, because hypophosphatemia has been associated with rhabdomyolysis in other states, administration of potassium phosphate in cases of severe hypophosphatemia may be considered for the purpose of trying to prevent rhabdomyolysis.
Complications
In Ontario, in-hospital mortality in people hospitalized for acute hyperglycemia ranged from <1% at ages 20 to 49 years to 16% in those over 75 years (73). Reported mortality in DKA ranges from 0.65% to 3.3% (3,39,74–76). In HHS, recent studies found mortality rates to be 12% to 17%, but included individuals with mixed DKA and hyperosmolality (2,5,77). About 50% of deaths occur in the first 48 to 72 hours. Mortality is usually due to the precipitating cause, electrolyte imbalances (especially hypo- and hyperkalemia) and cerebral edema.
Recommendations
-
In adults with DKA or HHS, a protocol should be followed that incorporates the following principles of treatment: fluid resuscitation, avoidance of hypokalemia, insulin administration, avoidance of rapidly falling serum osmolality and search for precipitating cause (as illustrated in Figure 1; see preamble for details of treatment for each condition) [Grade D, Consensus].
-
Point-of-care capillary beta-hydroxybutyrate may be measured in the hospital or outpatient setting [Grade D, Level 4 (33)] in adults with type 1 diabetes with CBG >14.0 mmol/L to screen for DKA, and a beta-hydroxybutyrate >1.5 mmol/L warrants further testing for DKA [Grade B, Level 2 (24–29)]. Negative urine ketones should not be used to rule out DKA [Grade D, Level 4 (35)].
-
In adults with DKA, intravenous 0.9% sodium chloride should be administered initially at 500 mL/h for 4 hours, then 250 mL/h for 4 hours [Grade B, Level 2 (50)] with consideration of a higher initial rate (1–2 L/h) in the presence of shock [Grade D, Consensus]. For adults with HHS, intravenous fluid administration should be individualized [Grade D, Consensus].
-
In adults with DKA, an infusion of short-acting intravenous insulin of 0.10 units/kg/h should be used [Grade B, Level 2 (54,55)]. The insulin infusion rate should be maintained until the resolution of ketosis [Grade B, Level 2 (60)] as measured by the normalization of the plasma anion gap [Grade D, Consensus]. Once the PG concentration falls to 14.0 mmol/L, intravenous dextrose should be started to avoid hypoglycemia [Grade D, Consensus].
-
Individuals treated with SGLT2 inhibitors with symptoms of DKA should be assessed for this condition even if BG is not elevated [Grade D, Consensus].
Abbreviations:
BG, blood glucose; CBG, capillary blood glucose; DKA, diabetic ketoacidosis; ECFV, extracellular fluid volume; HHS, hyperosmolar hyperglycemic state; KPD, ketosis-prone diabetes, PG, plasma glucose.
Other Relevant Guidelines
-
Glycemic Management in Adults With Type 1 Diabetes, p. S80
-
Pharmacologic Glycemic Management of Type 2 Diabetes in Adults, p. S88
-
Type 1 Diabetes in Children and Adolescents, p. S234
Relevant Appendix
- Appendix 8: Sick-Day Medication List
Literature Review Flow Diagram for Chapter 15: Hyperglycemic Emergencies in Adults
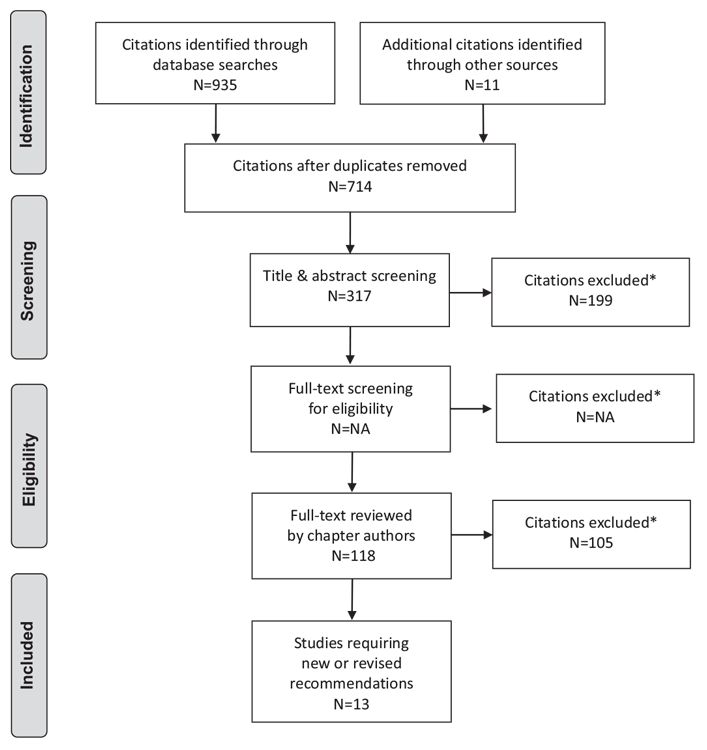
*Excluded based on: population, intervention/exposure, comparator/control or study design.
From: Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009). Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med 6(6): e1000097. doi:10.1371/journal.pmed1000097 (82).
For more information, visit www.prisma-statement.org.
Author Disclosures
Dr. Gilbert reports personal fees from Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Merck, Novo Nordisk, and Sanofi, outside the submitted work. Dr. Goguen does not have anything to disclose.
References
- Kitabchi AE, Umpierrez GE, Murphy MB, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care 2001;24:131–53.
- Hamblin PS, Topliss DJ, Chosich N, et al. Deaths associated with diabetic ketoacidosis and hyperosmolar coma. 1973–1988. Med J Aust 1989;151:41–2, 44.
- Holman RC, Herron CA, Sinnock P. Epidemiologic characteristics of mortality from diabetes with acidosis or coma, United States, 1970–78. Am J Public Health 1983;73:1169–73.
- Pasquel FJ, Umpierrez GE. Hyperosmolar hyperglycemic state: A historic review of the clinical presentation, diagnosis, and treatment. Diabetes Care 2014;37:3124–31.
- Wachtel TJ, Tetu-Mouradjian LM, Goldman DL, et al. Hyperosmolarity and acidosis in diabetes mellitus: A three-year experience in Rhode Island. J Gen Intern Med 1991;6:495–502.
- Malone ML, Gennis V, Goodwin JS. Characteristics of diabetic ketoacidosis in older versus younger adults. J Am Geriatr Soc 1992;40:1100–4.
- Wang ZH, Kihl-Selstam E, Eriksson JW. Ketoacidosis occurs in both type 1 and type 2 diabetes–a population-based study from Northern Sweden. Diabet Med 2008;25:867–70.
- Kitabchi AE, Umpierrez GE, Murphy MB, et al. Hyperglycemic crises in adult patients with diabetes: A consensus statement from the American Diabetes Association. Diabetes Care 2006;29:2739–48.
- Balasubramanyam A, Garza G, Rodriguez L, et al. Accuracy and predictive value of classification schemes for ketosis-prone diabetes. Diabetes Care 2006;29:2575–9.
- Laffel LM, Wentzell K, Loughlin C, et al. Sick day management using blood 3-hydroxybutyrate (3-OHB) compared with urine ketone monitoring reduces hospital visits in young people with T1DM: A randomized clinical trial. Diabet Med 2006;23:278–84.
- OgawaW, Sakaguchi K. Euglycemic diabetic ketoacidosis induced by SGLT2 inhibitors: Possible mechanism and contributing factors. J Diabetes Investig 2016;7:135–8.
- Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: A predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care 2015;38:1638–42.
- Singh AK. Sodium-glucose co-transporter-2 inhibitors and euglycemic ketoacidosis: Wisdom of hindsight. Indian J Endocrinol Metab 2015;19:722–30.
- Erondu N, Desai M, Ways K, et al. Diabetic ketoacidosis and related events in the canagliflozin type 2 diabetes clinical program. Diabetes Care 2015;38:1680–6.
- Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015;373:2117–28.
- Hayami T, Kato Y, Kamiya H, et al. Case of ketoacidosis by a sodium-glucose cotransporter 2 inhibitor in a diabetic patient with a low-carbohydrate diet. J Diabetes Investig 2015;6:587–90.
- Peters AL, Buschur EO, Buse JB, et al. Euglycemic diabetic ketoacidosis: A potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care 2015;38:1687–93.
- Redford C, Doherty L, Smith J. SGLT2 inhibitors and the risk of diabetic ketoacidosis. Practical Diabetes 2015;32:263–4.
- St Hilaire R, Costello H. Prescriber beware: Report of adverse effect of sodiumglucose cotransporter 2 inhibitor use in a patient with contraindication. Am J Emerg Med 2015;33:604, e3-.4.
- Goldenberg RM, Berard LD, Cheng AYY, et al. SGLT2 inhibitor-associated diabetic ketoacidosis: Clinical reviewand recommendations for prevention and diagnosis. Clin Ther 2016;38:2654–64, e1.
- Malatesha G, Singh NK, Bharija A, et al. Comparison of arterial and venous pH, bicarbonate, PCO2 and PO2 in initial emergency department assessment. Emerg Med J 2007;24:569–71.
- Brandenburg MA, Dire DJ. Comparison of arterial and venous blood gas values in the initial emergency department evaluation of patients with diabetic ketoacidosis. Ann Emerg Med 1998;31:459–65.
- Ma OJ, Rush MD, Godfrey MM, et al. Arterial blood gas results rarely influence emergency physician management of patients with suspected diabetic ketoacidosis. Acad Emerg Med 2003;10:836–41.
- Charles RA, Bee YM, Eng PH, et al. Point-of-care blood ketone testing: Screening for diabetic ketoacidosis at the emergency department. Singapore Med J 2007;48:986–9.
- Naunheim R, Jang TJ, Banet G, et al. Point-of-care test identifies diabetic ketoacidosis at triage. Acad Emerg Med 2006;13:683–5.
- Sefedini E, Prašek M, Metelko Z, et al. Use of capillary beta-hydroxybutyrate for the diagnosis of diabetic ketoacidosis at emergency room: Our one-year experience. Diabetol Croat 2008;37:73–80.
- Mackay L, Lyall MJ, Delaney S, et al. Are blood ketones a better predictor than urine ketones of acid base balance in diabetic ketoacidosis? Pract Diabetes Int 2010;27:396–9.
- Bektas F, Eray O, Sari R, et al. Point of care blood ketone testing of diabetic patients in the emergency department. Endocr Res 2004;30:395–402.
- Harris S, Ng R, Syed H, et al. Near patient blood ketone measurements and their utility in predicting diabetic ketoacidosis. Diabet Med 2005;22:221–4.
- Misra S, Oliver NS. Utility of ketone measurement in the prevention, diagnosis and management of diabetic ketoacidosis. Diabet Med 2015;32:14–23.
- Chiasson JL, Aris-Jilwan N, Belanger R, et al. Diagnosis and treatment of diabetic ketoacidosis and the hyperglycemic hyperosmolar state. CMAJ 2003;168:859–66.
- Lebovitz HE. Diabetic ketoacidosis. Lancet 1995;345:767–72.
- Cao X, Zhang X, Xian Y, et al. The diagnosis of diabetic acute complications using the glucose-ketone meter in outpatients at endocrinology department. Int J Clin Exp Med 2014;7:5701–5.
- Munro JF, Campbell IW, McCuish AC, et al. Euglycaemic diabetic ketoacidosis. Br Med J 1973;2:578–80.
- Kuru B, Sever M, Aksay E, et al. Comparing finger-stick beta-hydroxybutyrate with dipstick urine tests in the detection of ketone bodies. Turk J Emerg Med 2014;14:47–52.
- Guo RX, Yang LZ, Li LX, et al. Diabetic ketoacidosis in pregnancy tends to occur at lower blood glucose levels: Case-control study and a case report of euglycemic diabetic ketoacidosis in pregnancy. J Obstet Gynaecol Res 2008;34:324–30.
- Oliver R, Jagadeesan P, Howard RJ, et al. Euglycaemic diabetic ketoacidosis in pregnancy: An unusual presentation. J Obstet Gynaecol 2007;27:308.
- Chico A, Saigi I, Garcia-Patterson A, et al. Glycemic control and perinatal outcomes of pregnancies complicated by type 1 diabetes: Influence of continuous subcutaneous insulin infusion and lispro insulin. Diabetes Technol Ther 2010;12:937–45.
- May ME, Young C, King J. Resource utilization in treatment of diabetic ketoacidosis in adults. Am J Med Sci 1993;306:287–94.
- Levetan CS, Passaro MD, Jablonski KA, et al. Effect of physician specialty on outcomes in diabetic ketoacidosis. Diabetes Care 1999;22:1790–5.
- Ullal J, McFarland R, Bachand M, et al. Use of a computer-based insulin infusion algorithm to treat diabetic ketoacidosis in the emergency department. Diabetes Technol Ther 2016;18:100–3.
- Bull SV, Douglas IS, Foster M, et al. Mandatory protocol for treating adult patients with diabetic ketoacidosis decreases intensive care unit and hospital lengths of stay: Results of a nonrandomized trial. Crit Care Med 2007;35:41–6.
- Waller SL, Delaney S, Strachan MW. Does an integrated care pathway enhance the management of diabetic ketoacidosis? Diabet Med 2007;24:359–63.
- Devalia B. Adherance to protocol during the acutemanagement of diabetic ketoacidosis: Would specialist involvement lead to better outcomes? Int J Clin Pract 2010;64:1580–2.
- Salahuddin M, Anwar MN. Study on effectiveness of guidelines and high dependency unit management on diabetic ketoacidosis patients. J Postgrad Med Inst 2009;23:120–3.
- Corl DE, Yin TS, Mills ME, et al. Evaluation of point-of-care blood glucose measurements in patients with diabetic ketoacidosis or hyperglycemic hyperosmolar syndrome admitted to a critical care unit. J Diabetes Sci Technol 2013;7:1265–74.
- Kreisberg RA. Diabetic ketoacidosis: New concepts and trends in pathogenesis and treatment. Ann Intern Med 1978;88:681–95.
- Mahoney CP, Vlcek BW, DelAguila M. Risk factors for developing brain herniation during diabetic ketoacidosis. Pediatr Neurol 1999;21:721–7.
- Rosenbloom AL. Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes Care 1990;13:22–33.
- Adrogue HJ, Barrero J, Eknoyan G. Salutary effects of modest fluid replacement in the treatment of adults with diabetic ketoacidosis. Use in patients without extreme volume deficit. JAMA 1989;262:2108–13.
- Fein IA, Rachow EC, Sprung CL, et al. Relation of colloid osmotic pressure to arterial hypoxemia and cerebral edema during crystalloid volume loading of patients with diabetic ketoacidosis. Ann Intern Med 1982;96:570–5.
- Owen OE, Licht JH, Sapir DG. Renal function and effects of partial rehydration during diabetic ketoacidosis. Diabetes 1981;30:510–18.
- Kitabchi AE, Ayyagari V, Guerra SM. The efficacy of low-dose versus conventional therapy of insulin for treatment of diabetic ketoacidosis. Ann Intern Med 1976;84:633–8.
- Heber D, Molitch ME, Sperling MA. Low-dose continuous insulin therapy for diabetic ketoacidosis. Prospective comparison with “conventional” insulin therapy. Arch Intern Med 1977;137:1377–80.
- Butkiewicz EK, Leibson CL, O’Brien PC, et al. Insulin therapy for diabetic ketoacidosis. Bolus insulin injection versus continuous insulin infusion. Diabetes Care 1995;18:1187–90.
- Kitabchi AE, Murphy MB, Spencer J, et al. Is a priming dose of insulin necessary in a low-dose insulin protocol for the treatment of diabetic ketoacidosis? Diabetes Care 2008;31:2081–5.
- Fort P,Waters SM, Lifshitz F. Low-dose insulin infusion in the treatment of diabetic ketoacidosis: Bolus versus no bolus. J Pediatr 1980;96:36–40.
- Lindsay R, Bolte RG. The use of an insulin bolus in low-dose insulin infusion for pediatric diabetic ketoacidosis. Pediatr Emerg Care 1989;5:77–9.
- Andrade-Castellanos CA, Colunga-Lozano LE, Delgado-Figueroa N, et al. Subcutaneous rapid-acting insulin analogues for diabetic ketoacidosis. Cochrane Database Syst Rev 2016;(1):CD011281.
- Wiggam MI, O’Kane MJ, Harper R, et al. Treatment of diabetic ketoacidosis using normalization of blood 3-hydroxybutyrate concentration as the endpoint of emergencymanagement. A randomized controlled study. Diabetes Care 1997;20:1347–52.
- Morris LR, Murphy MB, Kitabchi AE. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med 1986;105:836–40.
- Gamba G, Oseguera J, Castrejón M, et al. Bicarbonate therapy in severe diabetic ketoacidosis. A double blind, randomized, placebo controlled trial. Rev Invest Clin 1991;43:234–8.
- Hale PJ, Crase J, Nattrass M. Metabolic effects of bicarbonate in the treatment of diabetic ketoacidosis. Br Med J (Clin Res Ed) 1984;289:1035–8.
- Soler NG, Bennett MA, Dixon K, et al. Potassium balance during treatment of diabetic ketoacidosis with special reference to the use of bicarbonate. Lancet 1972;2:665–7.
- Carlotti AP, Bohn D, Mallie JP, et al. Tonicity balance, and not electrolyte-free water calculations, more accurately guides therapy for acute changes in natremia. Intensive Care Med 2001;27:921–4.
- O’Malley G, Moran C, Draman MS, et al. Central pontine myelinolysis complicating treatment of the hyperglycaemic hyperosmolar state. Ann Clin Biochem 2008;45:440–3.
- Waldhausl W, Kleinberger G, Korn A, et al. Severe hyperglycemia: Effects of rehydration on endocrine derangements and blood glucose concentration. Diabetes 1979;28:577–84.
- Gerich JE, Martin MM, Recant L. Clinical and metabolic characteristics of hyperosmolar nonketotic coma. Diabetes 1971;20:228–38.
- Keller U, Berger W. Prevention of hypophosphatemia by phosphate infusion during treatment of diabetic ketoacidosis and hyperosmolar coma. Diabetes 1980;29:87–95.
- Wilson HK, Keuer SP, Lea AS, et al. Phosphate therapy in diabetic ketoacidosis. Arch Intern Med 1982;142:517–20.
- Fisher JN, Kitabchi AE. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab 1983;57:177–80.
- Singhal PC, Abramovici M, Ayer S, et al. Determinants of rhabdomyolysis in the diabetic state. Am J Nephrol 1991;11:447–50.
- Booth GL, Fang J. Acute complications of diabetes. In: Hux JE, Booth GL, Slaughter PM, et al., eds. Diabetes in Ontario: An iCES practice atlas. Toronto: Institute for Clinical Evaluative Science (ICES), 2003.
- Bagg W, Sathu A, Streat S, et al. Diabetic ketoacidosis in adults at Auckland hospital, 1988–1996. Aust N Z J Med 1998;28:604–8.
- Umpierrez GE, Kelly JP, Navarrete JE, et al. Hyperglycemic crises in urban blacks. Arch Intern Med 1997;157:669–75.
- Musey VC, Lee JK, Crawford R, et al. Diabetes in urban African-Americans. I. Cessation of insulin therapy is the major precipitating cause of diabetic ketoacidosis. Diabetes Care 1995;18:483–9.
- Wachtel TJ, Silliman RA, Lamberton P. Predisposing factors for the diabetic hyperosmolar state. Arch Intern Med 1987;147:499–501.
- Bellazzini MA, Meyer T. Pseudo-myocardial infarction in diabetic ketoacidosis with hyperkalemia. J Emerg Med 2010;39:e139–41.
- Petrov D, Petrov M. Widening of the QRS complex due to severe hyperkalemia as an acute complication of diabetic ketoacidosis. J Emerg Med 2008;34:459–61.
- Geddes J, Deans KA, Cormack A, et al. Cardiac troponin I concentrations in people presenting with diabetic ketoacidosis. Ann Clin Biochem 2007;44:391–3.
- Talapatra I, Tymms DJ. Diabetic ketoacidosis precipitated by subacute (De Quervain’s) thyroiditis. Pract Diabetes Int 2006;23:76–7.
- Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med 2009;6:e1000097.
Diabetes Canada is the registered owner of all content on guidelines.diabetes.ca and ShopDC. For questions, please email info@diabetes.ca